ev3密网支架召回~(解析国产器械鲜见召回背后的隐情)
来源:医休器械平台旗下公众号-医休神介说
作者:医休哥
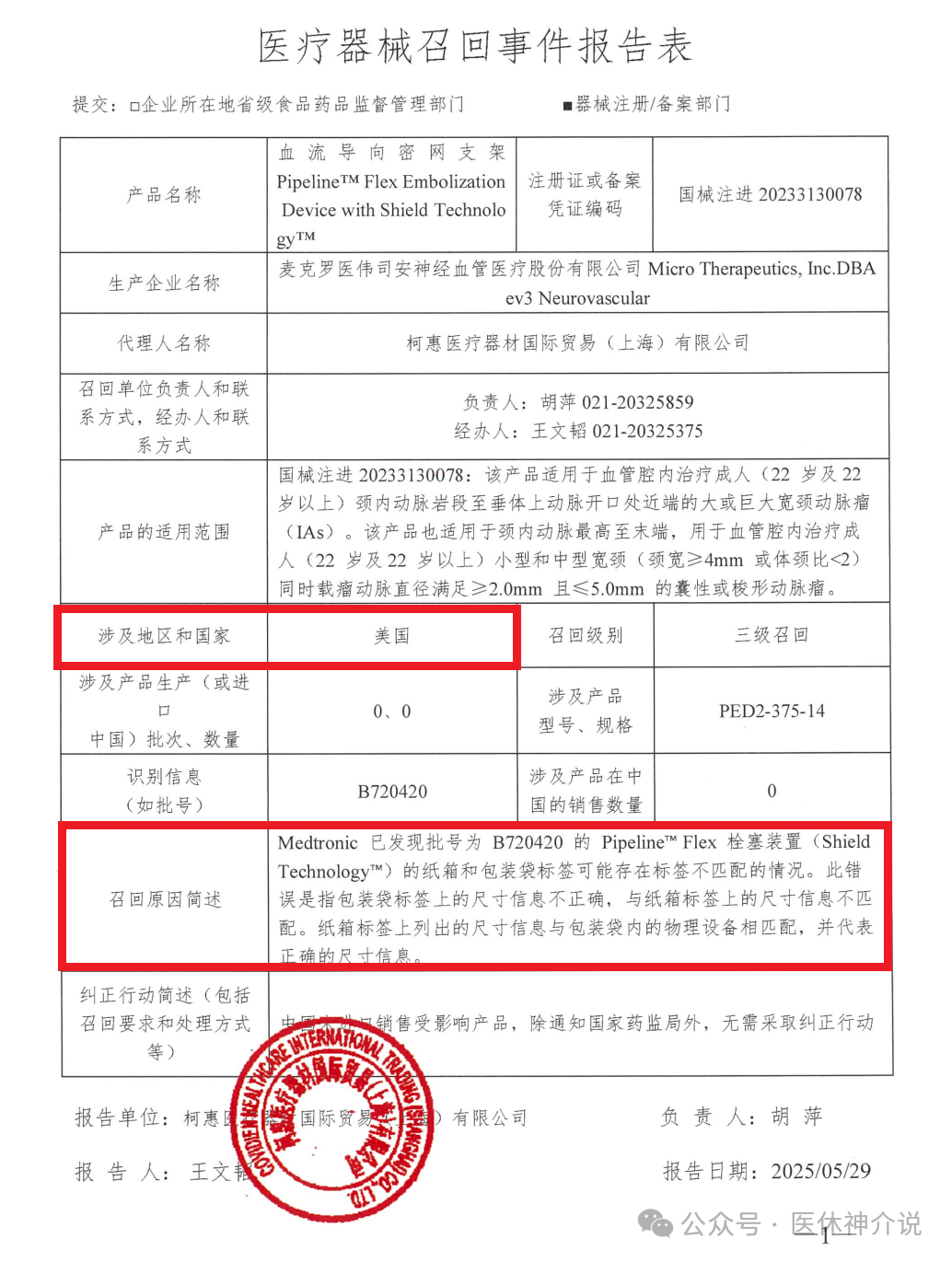
刚刚,国家药监局官网发布信息,柯惠医疗器材国际贸易(上海)有限公司报告,生产商麦克罗医伟司安神经血管医疗股份有限公司Micro Therapeutics,Inc.DBA ev3 Neurovascular对其生产的血流导向密网支架Pipeline Flex Embolization Device with Shield Technology(国械注进20233130078)主动召回。召回级别为三级召回。召回的主要原因是发现批号为B720420的PipelineFlex栓塞装置的纸箱和包装袋标签可能存在标签不匹配的情况。
本次召回涉及的产品未进口至中国,具体型号、规格及批次等详细信息见《医疗器械召回事件报告表》。

 可以看出,以上的召回问题不大,不过小编想和大家探讨的是,为何国内医疗器械召回情况很少见,而国外企业的召回事件却“很多”?
可以看出,以上的召回问题不大,不过小编想和大家探讨的是,为何国内医疗器械召回情况很少见,而国外企业的召回事件却“很多”?
FDA:大口径颅内抽吸导管Hippo因存在风险,被归类1级召回(最严重)
因包装不完整,Headway微导管主动召回~
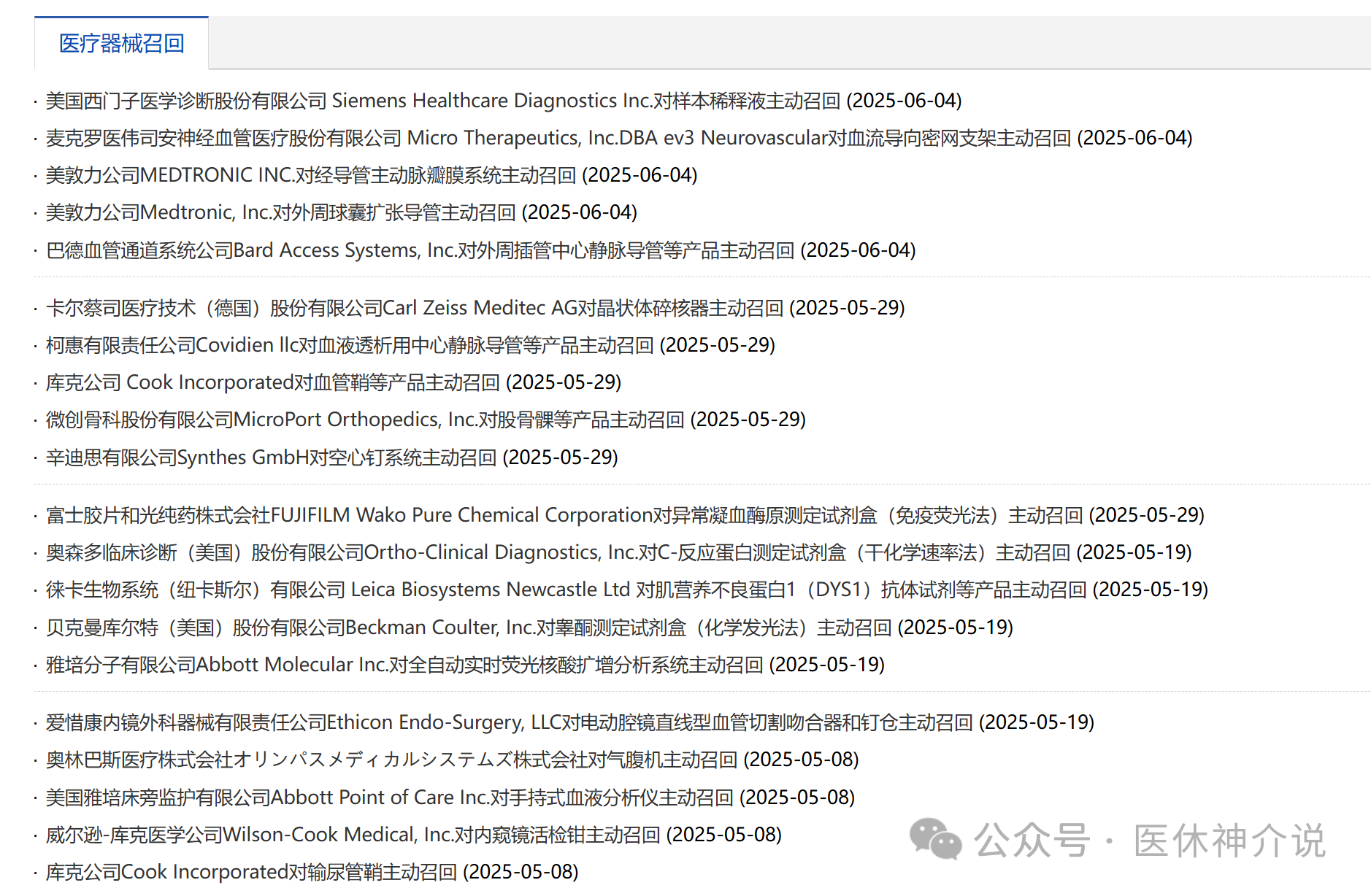
在中国,医疗器械召回的起步时间相比发达国家晚了近40年,召回制度始于美国。
1966年,美国汽车行业出台规定,制造商有义务召回存在潜在风险的汽车。1972年,他们颁布《消费品安全法案》,授权美国消费品安全委员会召回有潜在风险的产品,此举标志其产品召回制度正式确立。随后不久,该制度被引入药品监管领域。接下来的几十年里,欧美国家的产品召回制度日趋完善。
2011年7月1日,我国开始施行《医疗器械召回管理办法(试行)》,目的在于强化医疗器械的监督管理,保障人体健康和生命安全。这也是我国首部医疗器械召回法规。
2017年《医疗器械召回管理办法》正式颁布,局长是毕井泉(2025年5月29日,十四届全国政协常委、经济委员会副主任毕井泉因涉嫌严重违纪违法,被中央纪委国家监委纪律审查和监察调查。)
2001年10月,美国圣尤达心脏起搏器在中国召回,但当时国内尚无跟踪系统等基础支撑,使得召回工作举步维艰。这个教训引起有关管理部门的关注和反思。2002年全国“两会”期间,时任全国人大代表粱燕君等联名提交“建立完善缺陷产品召回制度”的建议。2002年4月23日,国家药监局发出限令,所有从疯牛病疫区进口的用牛羊组织为原料生产的医疗器械,都必须召回并禁止使用。
2005年,国家药监局处理医疗器械召回事件15起,涉及心脏除颤器、心脏起搏器、血糖仪及试纸等9个品种,其中13起由国外厂商发起。2007年12月6日,《药品召回管理办法》问世。
《医疗器械召回管理办法》的三个关键点
首先,界定了何谓医疗器械召回。
即医疗器械生产企业按照规定的程序对其已上市销售的存在缺陷的某一类别、型号或者批次的产品,采取警示、检查、修理、重新标签、修改并完善说明书、软件升级、替换、收回、销毁等方式消除缺陷的行为。
一些专家解读时,曾表达一个共同的意思:召回并不总是意味着必须立即停止使用该器械,或将该器械退回到制造商,有时仅仅表示一个医疗器械需要进行检查、调理或修理,甚至有时只是修改并完善说明书。因为“办法”依据医疗器械潜在风险的严重程度,将医疗器械召回分为3个级别:一级召回;二级召回;三级召回。级别数字越小,严重程度越高。
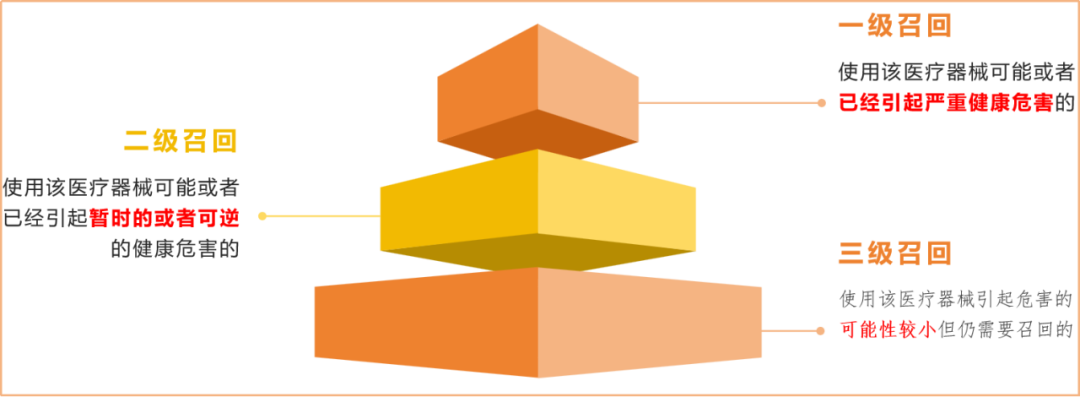
将召回分级管理,便是医疗器械召回法规的第二个关键点。
当然,有一种现象饱受诟病,曾有报道称个别外资医疗器械生产企业召回时,针对国外市场和中国市场采用双重标准——国外实施召回而国内迟迟无动于衷,抑或在我国降低召回级别。
第三个关键点则是基于启动召回的主体不同,将召回分为“主动召回”和“被动召回”。
前者简单来讲,即医疗器械生产商通过调查和评估,发现产品存在潜在风险,为防范伤害发生而主动采取预防性纠正措施。
与此相对应的是“责令召回”。也就是医疗器械生产企业应当召回医疗器械而未主动召回,药品监管部门向其送达责令召回通知书,强制企业召回具有潜在风险的产品的行政监管措施。
召回动力何在
医疗器械生产商为何愿意耗费巨资主动召回产品?
健康界获悉的一个解释是,欧美国家早已建立较为完善的医疗事故责任追溯制,若医疗事故是因医疗器械潜在风险引起的,生产企业被勒令支付的赔偿金额,要远远高于事故之前通过自查和主动召回而消除潜在风险的成本。
除了这个原因,他们之所以主动召回,还要归因于有产品召回险。比如美国,不少保险公司设立产品召回险,承保投保人的召回费用和第三方责任。
我国的《医疗器械召回管理办法(试行)》亦有敦促生产商主动召回的条款:医疗器械生产企业违反本办法规定,发现医疗器械存在缺陷而没有主动召回医疗器械的,责令召回医疗器械,并处应召回医疗器械货值金额3倍的罚款;造成严重后果的,由原发证部门吊销医疗器械产品注册证书,直至吊销《医疗器械生产企业许可证》。
国产器械少有召回,有何隐情?
·召回制度的强制性不足
2017年施行的《医疗器械召回管理办法》明确要求企业对缺陷产品主动召回,并设置三级召回时限(一级1日、二级3日、三级7日)。但企业违规成本相对较低:未主动召回的企业仅处货值金额3倍罚款,且“造成严重后果”才吊销许可证,威慑力有限。对比欧美,美国FDA对隐瞒缺陷可处以刑事处罚,欧盟MDR要求企业承担高额召回成本。
·责任主体界定模糊
法规虽明确境内注册人/备案人或进口代理人为召回责任主体,但中小型企业常因质量管理体系薄弱,难以有效追踪产品流向,导致召回执行困难。
·主动召回意愿较低声誉与市场顾虑
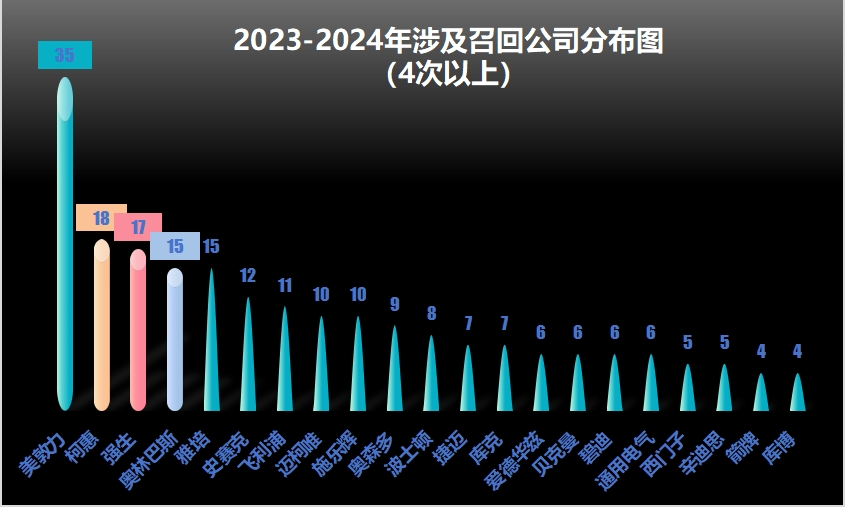
国内企业普遍担忧召回事件影响品牌形象和市场信任度。数据显示:77.9%的召回由企业自行发起,但进口器械占比75.1%,国产仅24.9%。
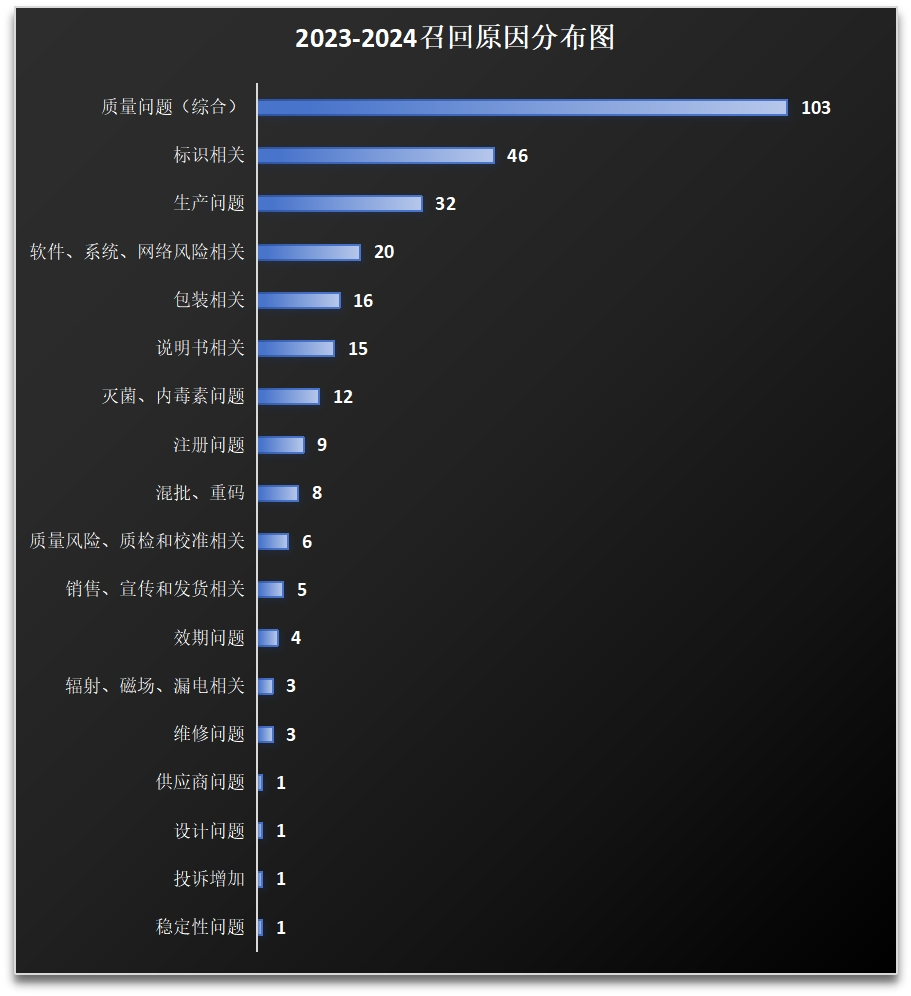
国产企业多依赖监管部门抽检(占比17.4%)或客户投诉(4.7%)后才被动召回,反映其风险自查动力不足。经济成本压力召回涉及物流、销毁、赔偿等费用,对利润空间有限的中小企业构成负担。例如:三级召回占比56.7%,多为标签错误等低风险问题,企业倾向通过“纠正措施”(如更换说明书)替代召回。一级召回仅占5.9%,且集中于设计缺陷(58.1%)等高风险问题,企业更倾向于采取私下补救措施而非公开召回。
许多企业对于召回持有顾虑,害怕商誉受损,因此即使在发现产品存在缺陷时,也往往选择私下处理而非公开召回。此外,一些企业对于召回的认识仍然存在误解,认为召回会伤害商业信誉,未能正确理解召回实际上是企业履行社会责任的表现。
·公众认知的不足
公众对医疗器械召回的重要性认识不足,也缺乏了解相关信息的渠道。同时,部分企业在召回过程中的信息公开不足,导致公众对召回事件的知晓度低。公众常将召回等同于“质量事故”,企业因此回避主动披露。上海市药监局曾指出需“加强宣传,提高社会对召回的接受度”。反观国际跨国企业通过主动召回塑造“负责任”形象,但国内企业尚未形成此类文化。
·供应链复杂性阻碍追溯
医疗器械流通环节多(生产→分销→医院→患者),企业难以及时获取终端使用数据。国产企业召回多依赖医院反馈,但医疗机构不良事件上报率不足30%。
本文2025-06-23 13:38:04发表“医休观点”栏目。
本文链接:https://www.yixiuqixie.com/article/825.html
相关文章



- 脑机新声-中国脑机接口产业14城城市布局深度研究报告202605.pdf
- 全球非侵入式脑机接口(BCI)行业深度调研与代表性企业进展报告.pdf
- 脑机新声内参:全球脑机接口电极与核心材料技术全景解析.pdf
- 脑机新声内参(下册):中国脑机接口产业扶持与宏观政策汇编.pdf
- 脑机新声汇编《中国脑机接口医疗准入与医保定价全景汇编(上册)》.pdf
- 脑机接口入行指导手册-脑机新声0505(1).pdf
- 河北省脑机接口产业发展规划建议-脑机新声.pdf
- 传统药企进入脑机接口赛道行业研究及可行性分析报告.pdf
- 【压缩版】神经介入企业进入脑机接口赛道的机会和挑战.pdf
- 【脑机新声】全球脑机接口技术前沿:2026年十大最具代表性BCI公司临床进展与行业深度洞察报告.pdf